| tags: [ Data Cleaning Genomic Data Linux PLINK R ] categories: [Coding Experiments ]
Cleaning and preparing genomic data
Introduction
After performing basic stats and QC on the genomic data sets, merging, and subsetting the data for NIES individuals, a few issues came to my attention. Firstly, from the histograms of the distribution of allele frequencies from the whole genome sequencing data, it became evident that the data had been filtered to exlude SNPs with MAF < 0.05. Thus any rare variants that I need for GWAS was excluded. Second, the IDs in the whole genome sequencing data were not UUIDs. Instead they were IDs that were generated and used in the lab. This means that the merging and subsetting I performed did not include any WGS data and most likely included individuals that may not have been part of the NIES. I have since received the unfiltered WGS data and an updated spreadsheet of study individuals and will re-do the cleaning and prep steps for the genomic data prior to performing PCA.
Methods
1. Discard all outputs from PLINK and old WGS file sets.
2. Download new WGS file set
3. Find allele frequencies in SNP and WGS file sets
plink1.9 --bfile NI_wgs_merged_snps --freq --out plink_output/wgs_allele_freqOutput: - 19,208,586 variants - 108 individuals (58 males, 50 females) - 108 founders - 2,739,848 variants with heterozygous haploid genotype present (warning) - genotyping rate is 99.68%
plink1.9 --bfile NImerged.impute2.chrAllX.2014-Oct-02 --freq --out plink_output/snp_allele_freqOutput: - 27,310,409 variants - 506 individuals (247 males, 259 females) - 45 founders present - genotyping rate is 98.65%
4. Generate histogram for allele frequency distributions for WGS data.
wgs_allele_freq <- read.table('C:/Users/Martha/Documents/Honours/Project/honours.project/Data/plink_output/wgs_allele_freq.frq', header = T)
hist(wgs_allele_freq$MAF, main = "Allele Frequency Distributions of WGS Data", xlab = "Minor Allele Frequency", xlim = c(0.0, 0.5), col = "darkseagreen1")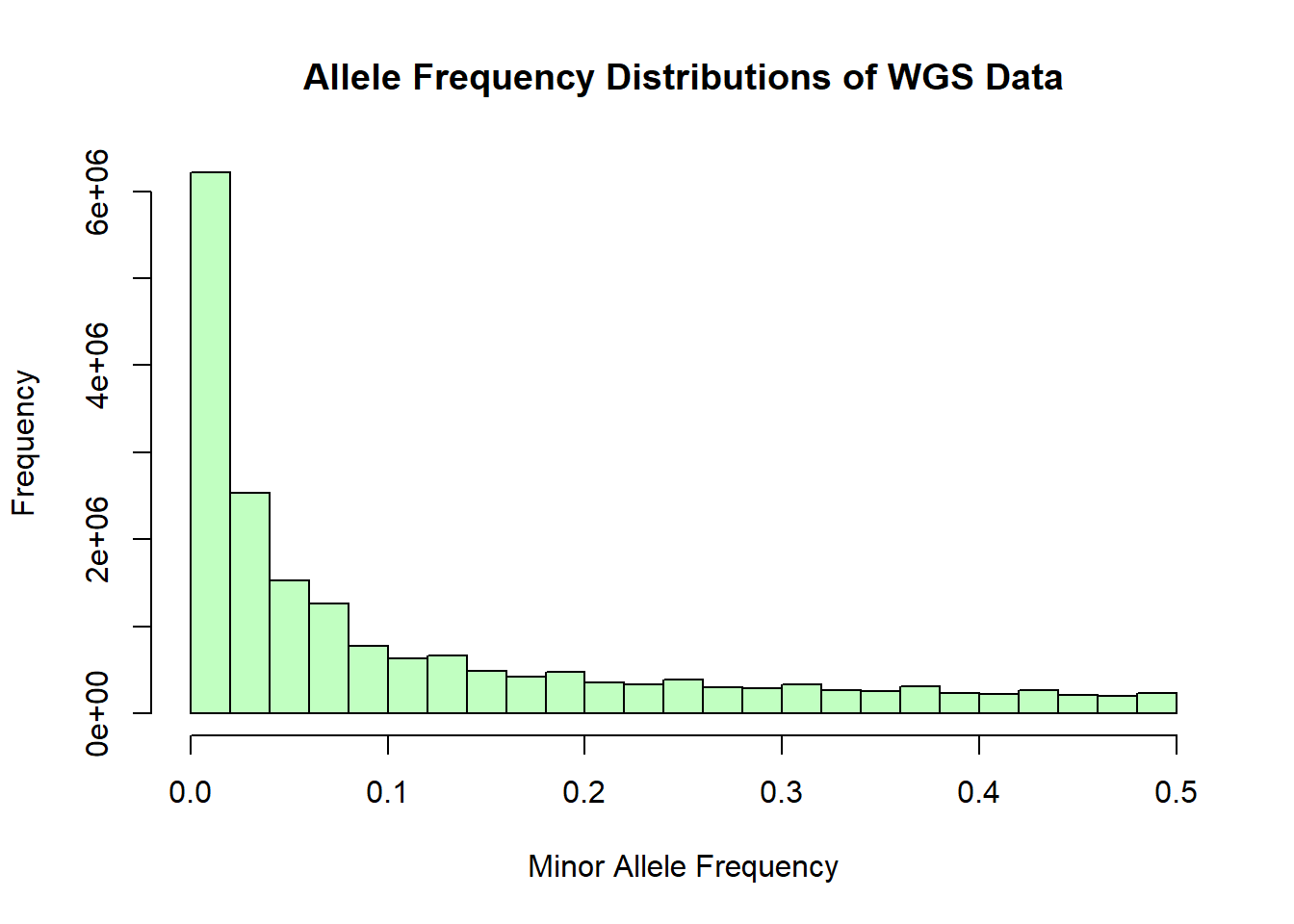
5. Generate histogram for allele frequency distributions for SNP data.
snp_allele_freq <- read.table('C:/Users/Martha/Documents/Honours/Project/honours.project/Data/plink_output/snp_allele_freq.frq', header = T)
hist(snp_allele_freq$MAF, main = "Allele Frequency Distribution of SNP Data", xlab = "Minor Allele Frequency", xlim = c(0.0, 0.5), col = "cadetblue1")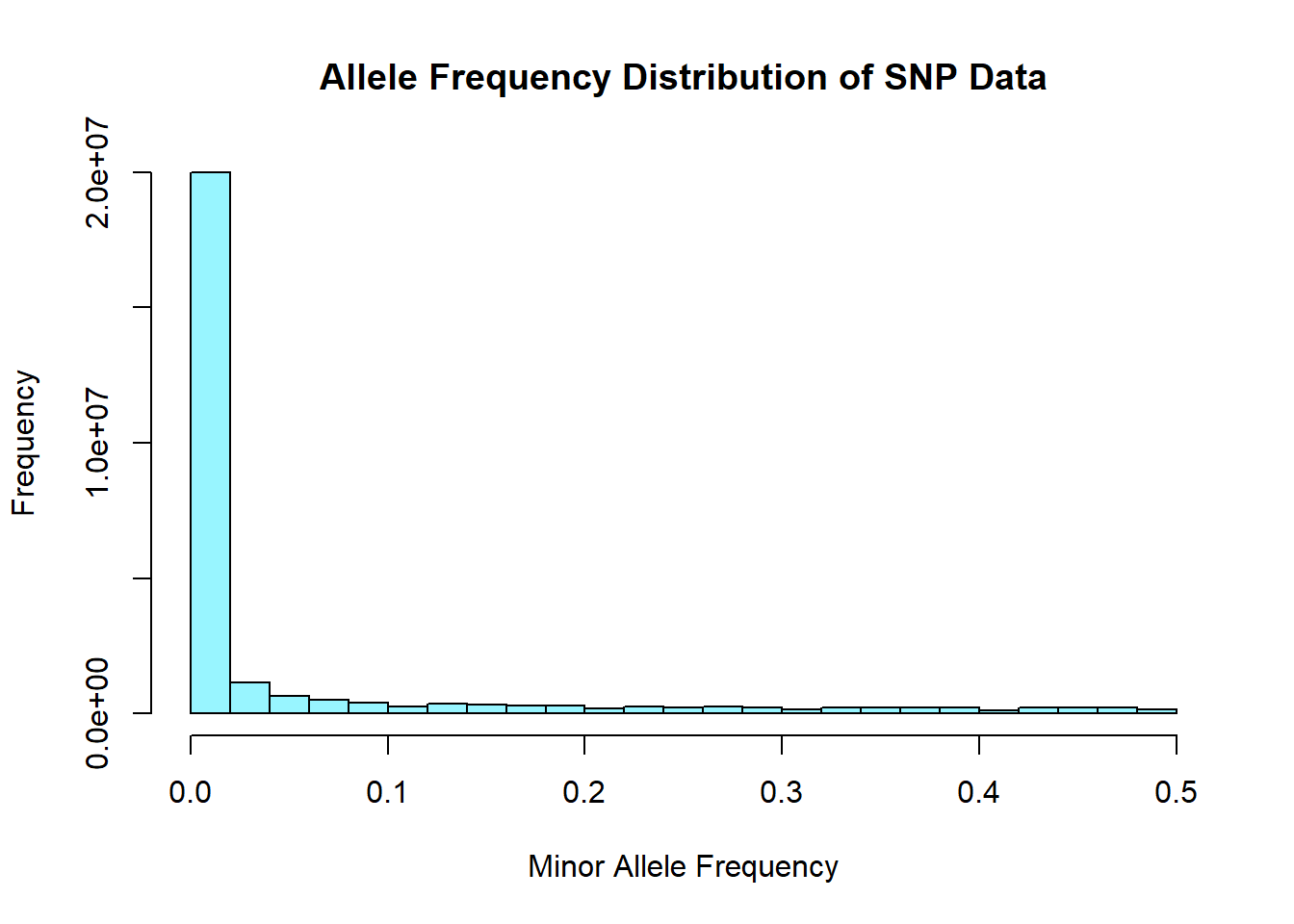
6. Find missingness for both data sets
plink1.9 --bfile NI_wgs_merged_snps --missing --out plink_output/wgs_missing
plink1.9 --bfile NImerged.impute2.chrAllX.2014-Oct-02 --missing --out plink_output/snp_missing7. Generate Hardy-Weinberg test statistics
plink1.9 --bfile NI_wgs_merged_snps --hardy --out plink_output/wgs_hardy
plink1.9 --bfile NImerged.impute2.chrAllX.2014-Oct-02 --hardy --out plink_output/snp_hardy8. Filter out variants based on HWE p-value
plink1.9 --bfile NI_wgs_merged_snps --hwe 1.8e-7 --make-bed --out plink_output/wgs_hwe_filterOutput: - Warning: hwe observation counts vary by more than 10% - 305,519 variants removed - 18,903,067 variants remain
plink1.9 --bfile NImerged.impute2.chrAllX.2014-Oct-02 --hwe 1.8e-7 --make-bed --out plink_output/snp_hwe_filterOutput: - Warning: hwe observation counts vary by more than 10% - 18 variants removed - 27,310,391 variants remain
NOTE: Filtering out based on HWE test created new file sets.
9. Merge data sets to identify variants with 3+ alleles
plink1.9 --bfile plink_output/wgs_hwe_filter --bmerge plink_output/snp_hwe_filter.bed plink_output/snp_hwe_filter.bim plink_output/snp_hwe_filter.fam --make-bed --out plink_output/trial_mergeOutput: - 18,903,067 variants loaded from WGS data, 27,310,391 variants loaded from SNP data - 17,865,008 variants are uncommon - 9,445,383 variants are common in both file sets - 9,362,232 variants have multiple position warnings - 11,641 variants have 3+ allele and caused merge error
10. Exclude variants with 3+ alleles from both file sets
plink1.9 --bfile plink_output/wgs_hwe_filter --exclude plink_output/trial_merge-merge.missnp --make-bed --out plink_output/wgs_excludeOutput (WGS): - 18,891,435 variants remain after excluding variants with 3+ alleles - genotyping rate is 99.69%
plink1.9 --bfile plink_output/snp_hwe_filter --exclude plink_output/trial_merge-merge.missnp --make-bed --out plink_output/snp_excludeOutput (SNP): - 27,298,645 variants remain after excluding variants with 3+ alleles - genotyping rate is 98.65%
NOTE: This step creates a new file set for WGS and SNP data
11. Re-try file merge
plink1.9 --bfile plink_output/wgs_exclude --bmerge plink_output/snp_exclude.bed plink_output/snp_exclude.bim plink_output/snp_exclude.fam --make-bed --out plink_output/var_out_mergeThis merge failed. 16GB of RAM on my laptop is not adequate for this function.