| tags: [ Data Cleaning Genomic Data PLINK R ] categories: [Coding Experiments ]
Troubleshooting missing genotype filtering
Introduction
Applying a missing genotype filter for individuals and markers (described in previous entry) yielded unexpected results as a significant number of individuals and variants were removed. Considering the high genotyping rates (~99%) of the original files (WGS and SNP-array), there should only be a small number of variants removed due to missing genotypes.
This exercise is aimed at determining the possible cause for this filtering issue by testing the filtering parameters on the original data set and merged data set. This should identify the step at which the genotyping rate changes.
Note: this exercise was performed on taurus #Methods and Results ###1. Apply missingness filters on original WGS data set
plink --bfile NI_wgs_merged_snps --mind 0.05 --make-bed -out test_output/wgs_imiss_filterOutput: No individuals were removed.
plink --bfile NI_wgs_merged_snps --geno 0.1 --make-bed -out test_output/wgs_imiss_filterOutput: 1 variant removed
plink --bfile NI_wgs_merged_snps --geno 0.01 --make-bed --out test_output/wgs_imiss_filterOutput: 781,724 variants removed (18,426,862 variants remain)
2. Apply missingness filters on original SNP-array data set
plink --bfile NI.snp.hg38_final --mind 0.05 --make-bed --out test_output/array_imiss_filterOutput: No individuals were removed.
plink --bfile NI.snp.hg38_final --geno 0.1 --make-bed -out test_output/array_imiss_filteroutput: 810,752 variants removed (25,244,548 variants remain)
plink --bfile NI.snp.hg38_final --geno 0.01 --make-bed --out test_output/array_imiss_filterOutput: 4,554,230 variants removed (21,501,070 variants remain)
3. Apply missingness filters on merged data set
plink --bfile plink_output/filtered_merge --mind 0.05 --make-bed --out test_output/merged_miss_filterOutput: 96 individuals removed (424 individuals remain)
plink --bfile plink_output/filtered_merge --mind 0.1 --make-bed --out test_output/merged_miss_filterOutput: 94 individuals removed (426 individuals remain)
plink --bfile plink_output/filtered_merge --geno 0.01 --make-bed --out test_output/merged_miss_filterOutput: 8,003,914 variants removed (1,151,139 variants remain)
plink --bfile plink_output/filtered_merge --geno 0.1 --make-bed --out test_output/merged_miss_filterOutput: 2,857,039 variants removed (6,298,014 variants remain)
Applying the filtering parameters on the merged data set indicates that the change in the genotyping rates occurred because of this merge. From the PLINK manual (http://zzz.bwh.harvard.edu/plink/dataman.shtml#merge), it appears that the default merge mode uses a consensus. This means that if there is a missing genotype in one file, it is overwritten by the other file. However, if a genotype is present in both files but they mismatch, it is treated as missing.
4. Re-run merge-mode 6 to determine mismatching calls between the two files.
plink --bfile plink_output/wgs_filtered --bmerge plink_output/array_filtered.bed plink_output/array_filtered.bim plink_output/array_filtered.fam --merge-mode 6 --out plink_output/merged_mode6_filteredOutput: - 860,574,982 overlapping calls - 835,821,129 non-missing in both filesets - 579,288,999 concordant - 69.31% concordance rate
This low concordance rate indicates that there is a substantial number of calls between the file sets that mismatch. Therefore, merging them using the default setting would identify these calls as missing, which explains the large number of individuals and variants filtered out.
5. Use merge-mode 3 to merge the file sets.
plink --bfile plink_output/array_filtered --bmerge plink_output/wgs_filtered.bed plink_output/wgs_filtered.bim plink_output/wgs_filtered.fam --merge-mode 3 --make-bed --out plink_output/filtered_merge_mode3Output: - 9,155,053 variants and 520 individuals pass filters and QC - total genotyping rate = 97.7%
Already, this total genotyping rate has increased compared to using the default merge setting (~92%).
6. Generate allele frequency stats
plink1.9 --bfile plink_output/filtered_merge_mode3 --freq --out plink_output/merged_mode3_freq7. Load allele freq file
merged_mode3_freq <- read.table('C:/Users/Martha/Documents/Honours/Project/honours.project/Data/plink_output/merged_mode3_freq.frq', header = T)8. Generate histogram for allele frequency distributions
hist(merged_mode3_freq$MAF, main = "Allele Frequency Distributions of Merged Data Set (Mode3)", xlab = "Minor Allele Frequency", xlim = c(0.0, 0.5), col = "turquoise3")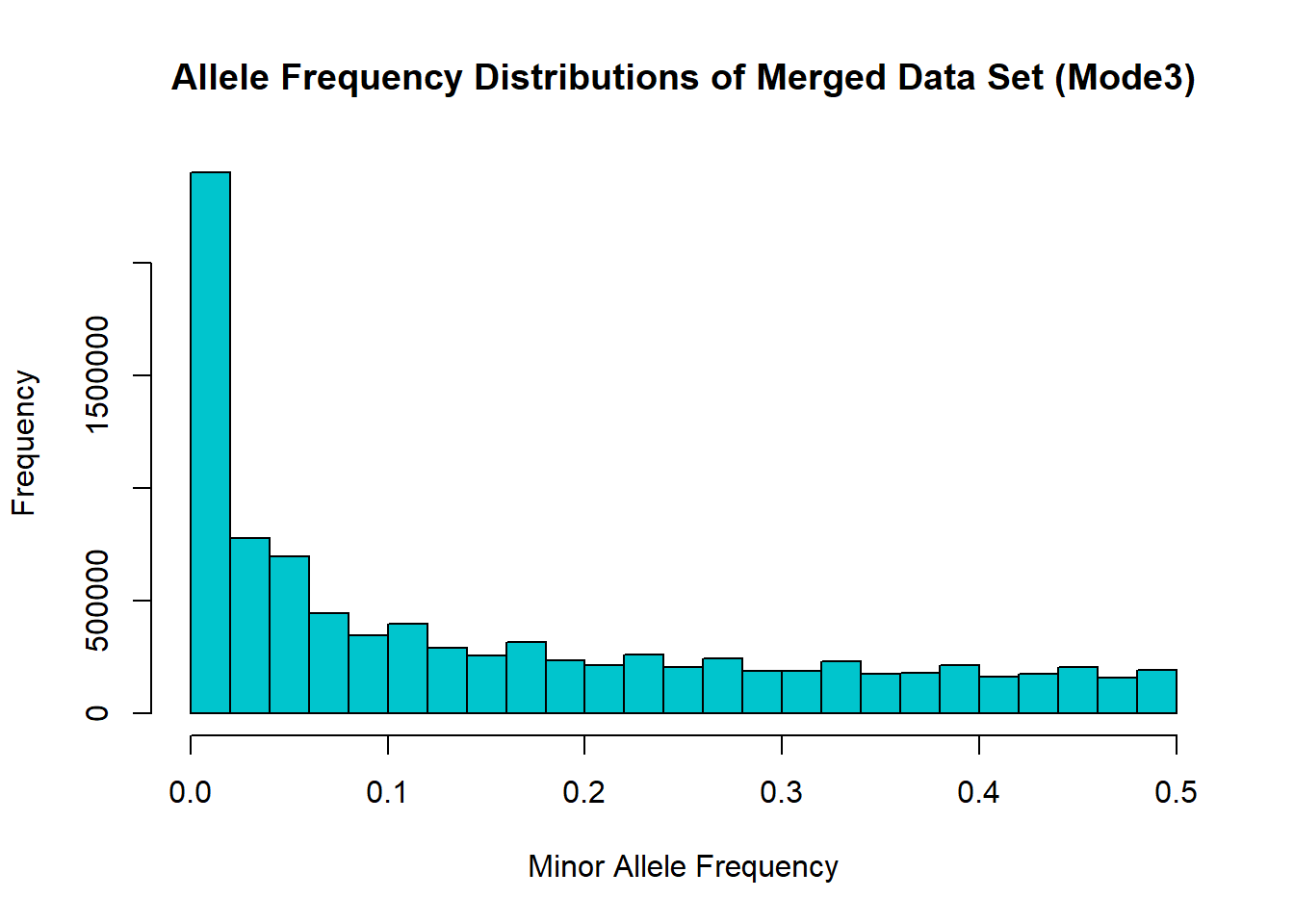
9. Filter out variants based on HWE p-value
plink1.9 --bfile plink_output/filtered_merge_mode3 --hwe 1.8e-7 --make-bed --out plink_output/merged_hwe_filterOutput: - 2295 variants removed due to HWE p-value - 9,152,758 variants remain
10. Load spreadsheet with NI study participants
ni_ped_uuid <- read.csv('C:/Users/Martha/Documents/Honours/Project/honours.project/Data/NI_UUID_Ped_20180718.csv', header = T)
head(ni_ped_uuid)## UUID Ped_ID LAB_ID PID SID NIES KCCGID Gender DOB IN_2000
## 1 110020 NA 1002 <NA> <NA> <NA> <NA> Female 5/16/1929 1
## 2 110030 NA 1003 <NA> <NA> <NA> <NA> Female 5/23/1928 1
## 3 110040 3676 1004 <NA> <NA> <NA> <NA> Female 2/08/1959 1
## 4 110050 765 1005 <NA> <NA> <NA> <NA> Male 11/07/1954 1
## 5 110080 NA 1008 <NA> <NA> <NA> <NA> Female 10/07/1933 1
## 6 110090 NA 1009 <NA> <NA> <NA> <NA> Female 10/08/1968 1
## AGE_2000NIHS IN_2010 AGE_2010NIHS IN_NIES AGE_NIES RECOLLECT COREPED_1
## 1 71 0 80 0 79 0 0
## 2 72 0 81 0 80 0 0
## 3 41 0 50 0 49 0 0
## 4 46 0 55 0 54 0 1
## 5 67 0 76 0 75 0 0
## 6 32 0 41 0 40 0 0
## COREPED_2 Miles_core DNA_NIHS X2000NIHS_GWAS X2010NIHS_GWAS GWAS_NIHS
## 1 0 0 1 0 0 0
## 2 0 0 1 0 0 0
## 3 0 0 1 0 0 0
## 4 1 1 1 1 0 1
## 5 0 0 1 0 0 0
## 6 0 0 1 0 0 0
## DNA_NIES GWAS_NIES Total_GWAS NI_exprs WGS_data DNA_TOTAL
## 1 0 0 0 0 0 1
## 2 0 0 0 0 0 1
## 3 0 0 0 0 0 1
## 4 0 0 1 0 0 1
## 5 0 0 0 0 0 1
## 6 0 0 0 0 0 1
## Relation.to.NIHS.Pedigree..n.6379. patID matID Ped_Gender isParent
## 1 <NA> NA NA <NA> NA
## 2 <NA> NA NA <NA> NA
## 3 <NA> NA NA <NA> NA
## 4 <NA> 764 762 Male FALSE
## 5 <NA> NA NA <NA> NA
## 6 <NA> NA NA <NA> NA11. Isolate NIES individuals using “NIES” column, then all NIES individuals with sequencing data using “GWAS_NIES” column
gwas_niesID <- read.csv('C:/Users/Martha/Documents/Honours/Project/honours.project/Data/gwas_niesID.csv', header = F)
head(gwas_niesID)## V1
## 1 312481
## 2 311020
## 3 255711
## 4 321130
## 5 232110
## 6 25381012. Extract data in PLINK
plink1.9 --bfile plink_output/merged_hwe_filter --keep gwas_niesID.txt --make-bed --out merged_niesOutput: - 361 people and 9,152,758 variants remain - total genotyping rate = 97.68%
13. Check allele freq stats for this data set
plink1.9 --bfile merged_nies --freq --out plink_output/merged_nies_allele_freq14. Load allele frequency file
merged_nies_allele_freq <- read.table('C:/Users/Martha/Documents/Honours/Project/honours.project/Data/plink_output/merged_nies_allele_freq.frq', header = T)15. Generate histogram for allele frequency distribution
hist(merged_nies_allele_freq$MAF, main = "Allele Frequency Distributions of NIES Data", xlab = "Minor Allele Frequency", xlim = c(0.0, 0.5), col = "lightskyblue1")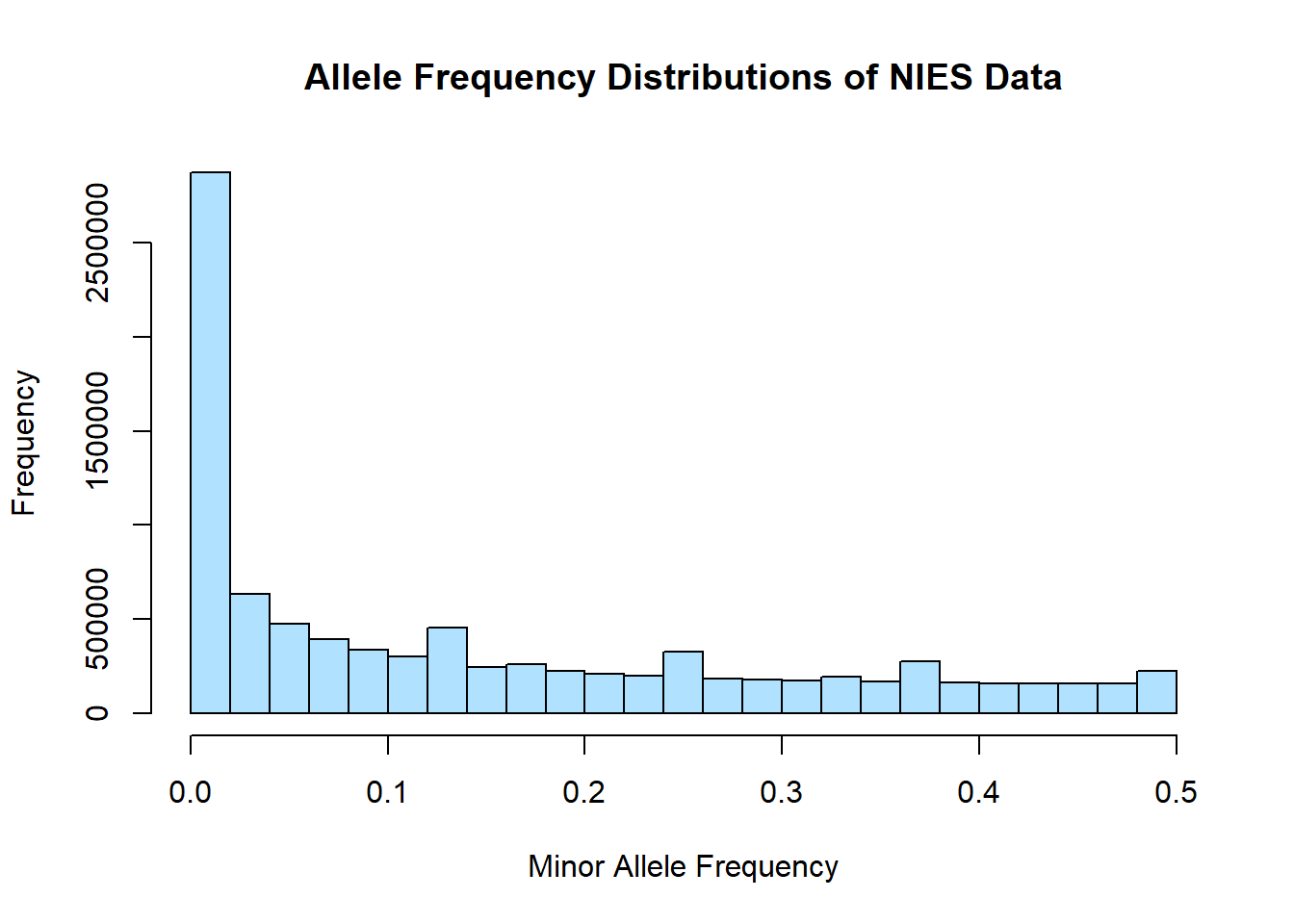
16. Apply filter for missing genotypes
plink1.9 --bfile merged_nies --mind 0.05 --geno 0.01 --make-bed --out nies_miss_filteredOutput: - 2 individuals removed (359 individuals remain) - 3,160,490 variants removed (5,992,268 variants remain) - total genotyping rate = 97.70%
Although this has significantly improved, two individuals were still removed and there is still a substantial number of variants being filtered out.
17. Apply a less stringent filter
plink1.9 --bfile merged_nies --mind 0.1 --geno 0.05 --make-bed --out nies_miss_filteredOutput: - 0 individuals removed - 1,096,020 variants removed (8,056,738 variants remain)
18. Check allele frequency
plink1.9 --bfile nies_miss_filtered --freq --out plink_output/nies_merged_snp_freqOutput: - 361 individuals loaded - 8,056,738 variants - total genotyping rate = 99.24%
19. Load allele frequency file
nies_merged_snp_freq <- read.table('C:/Users/Martha/Documents/Honours/Project/honours.project/Data/plink_output/nies_merged_snp_freq.frq', header = T)20. Generate histogram for allele frequency distribution
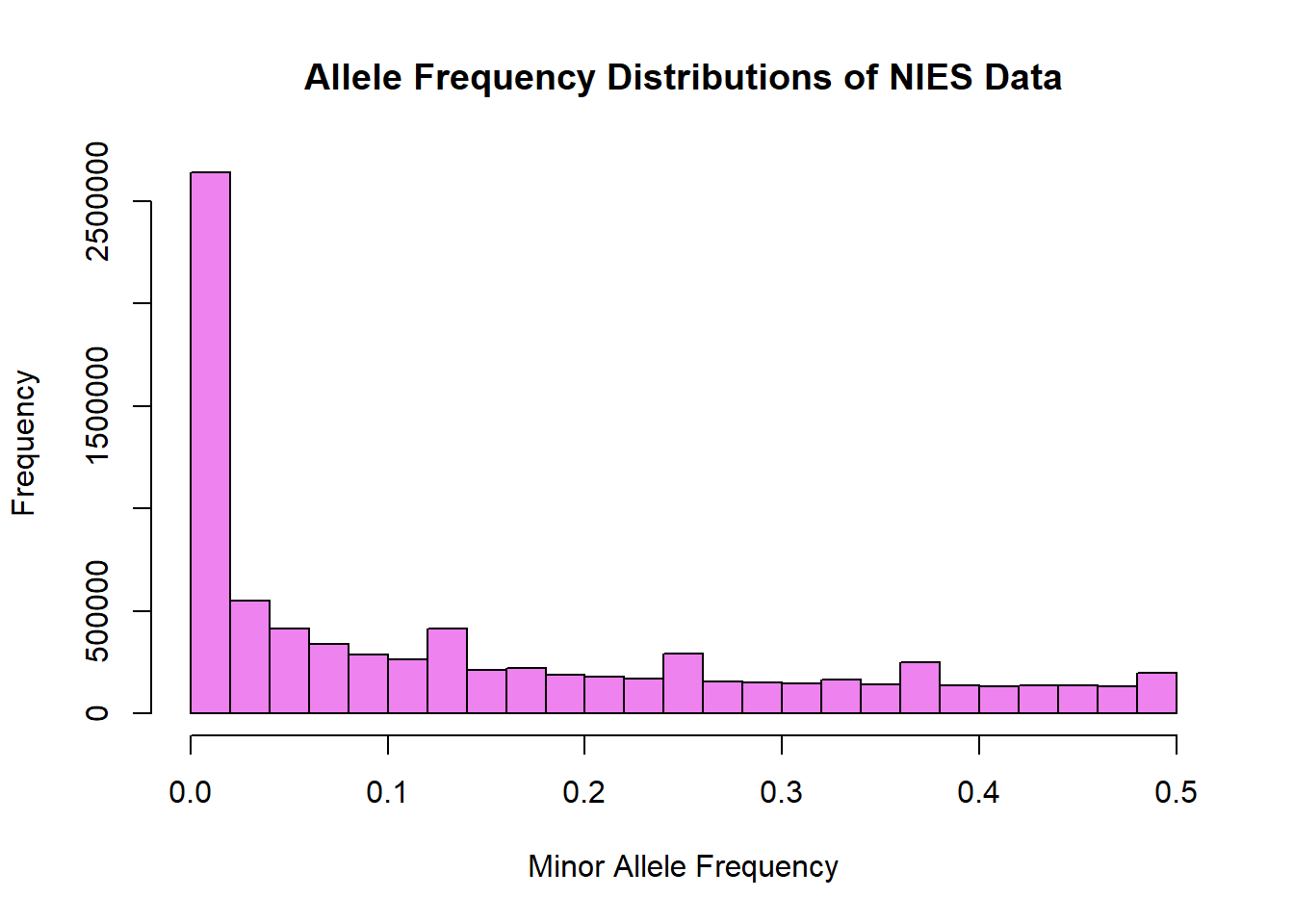
hist(nies_merged_snp_freq$MAF, main = "Allele Frequency Distributions of NIES Data", xlab = "Minor Allele Frequency", xlim = c(0.0, 0.5), col = "violet")